President Obama just signed the Food and Drug Administration Safety and Innovation Act. The Act includes the third authorisation of user fees, paid by the medical-device industry to the FDA, so that the FDA can improve its performance in licensing new medical devices.

The FDAs process has long been unsatisfactory. This is why the medical-device industry initially accepted the policy of paying user fees to the FDA, starting in 2003. These user fees increased the budget available for reviewing applications, above and beyond that which was appropriated by Congress from taxpayers. In return, the FDA committed to meeting certain targets for reviewing applications for new medical devices in a timely and transparent way.
Unfortunately, user fees for medical devices have had mixed results. Although they have expanded the FDAs budget, the FDAs regulatory processes have become less productive, according to an analysis I recently conducted for the Pacific Research Institute, available here.
U.S. law defines three classes of risk for medical devices. Class I devices (for example, adhesive tape or bedpans) pose minimal risk; Class II (for example, syringes or hearing aids) pose median risk; and Class III (for example, pacemakers and heart valves) pose the highest risk. Most Class I devices are exempt from prior approval; whereas 80 percent of Class II products are approved via the so-called 510(k) system; and Class III devices must go through a much more rigorous process called Premarket Approval (PMA).
Medical devices in use in the U.S. are remarkably safe. An expert review of higest-risk safety recalls over the six years from 2005 through 2009 concluded that over 99.5 percent of both 510(k) submissions and PMA submissions during this period did not result in a recall. So, the FDA is not being slack in approving medical devices without adequate oversight.
And yet, there are still widely recognized problems with the FDAs ability to allow new medicines onto the market in a timely and effective manner. Last summer, the prestigious Institute of Medicine (IOM) published the report of an expert committee, which concluded that: It is unclear and the committee concludes that it is indeterminable, given current information whether the 510(k) process over the last 35 years has had a positive or negative effect on innovation.
The FDA must change the way it uses its ever increasing funding for medical-device regulation. In the three years before user fees were introduced (2000 through 2002), the average annual number of 510(k) submissions was 4,300. The average annual number of PMA submissions was 77. The figures for the first five years of user fees (2003 through 2007) were down to 3,869 and 55; and for the next four years after the 2007 reauthorization (2008 through 2011) they were only 3,979 and 48.
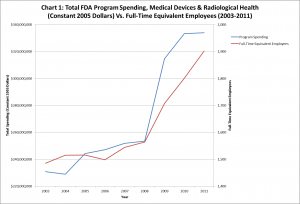
However, the amount of money spent on medical-device regulation increased dramatically, from $217 million in 2003 to over $378 million in 2011 an increase of three quarters. Even accounting for inflation, the increase in funding was 45 percent just under half over the period. Furthermore, the number of full-time equivalent employees at the FDA who are dedicated to medical-device regulation increased from 1,485 to 1,902, an increase of over one quarter. Both these data series are shown in Chart 1. (Click to expand.)
Americas lead in medical innovation is threatened by this decline in productivity. If we want the medical-device industry to keep investing and innovating in the United States, Americans will have to demand significant improvement in the FDAs regulation of medical devices.
Source: https://www.forbes.com/sites/aroy/2012/07/12/user-fees-for-medical-devices-third-time-lucky/